goal: deconvolute the NIAD 20 mix
================================================================
SUMMARY:
- I ran the "20" mixtures and got about 3000 full length reads per run
- Here are the clusters:
6MIX
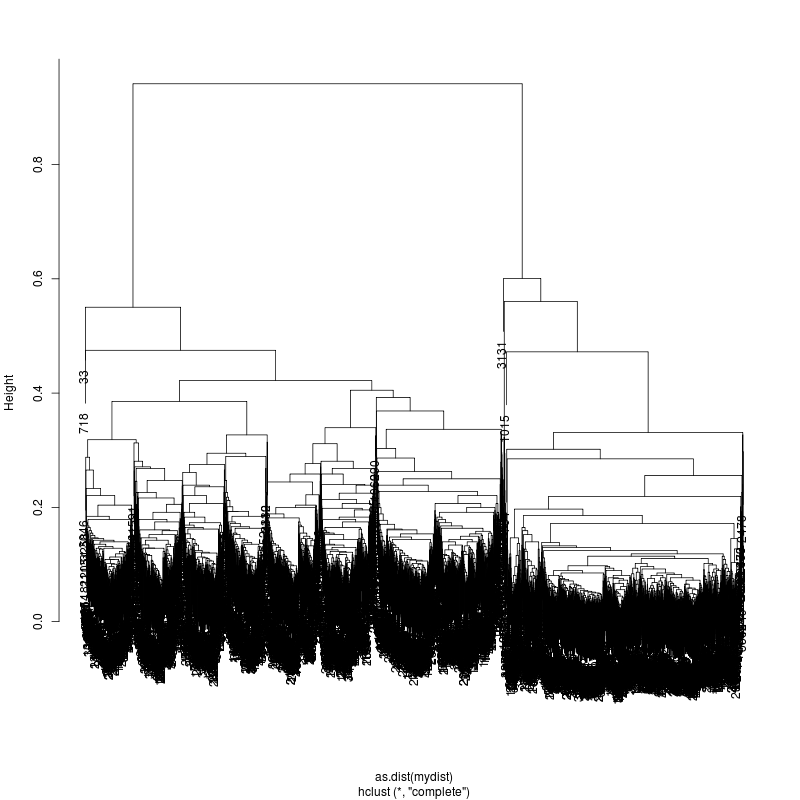
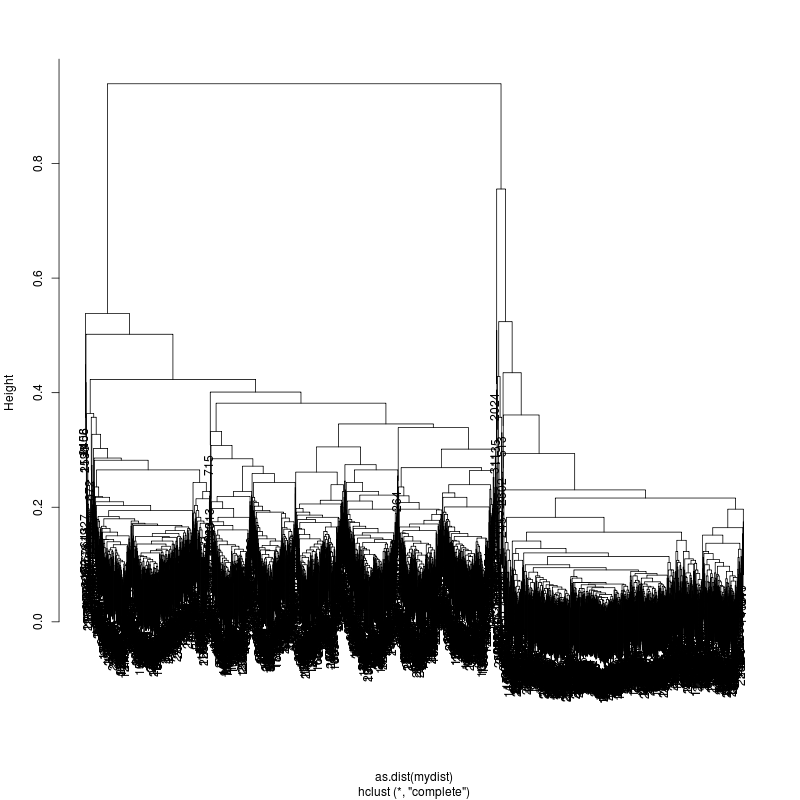 13MIX
13MIX
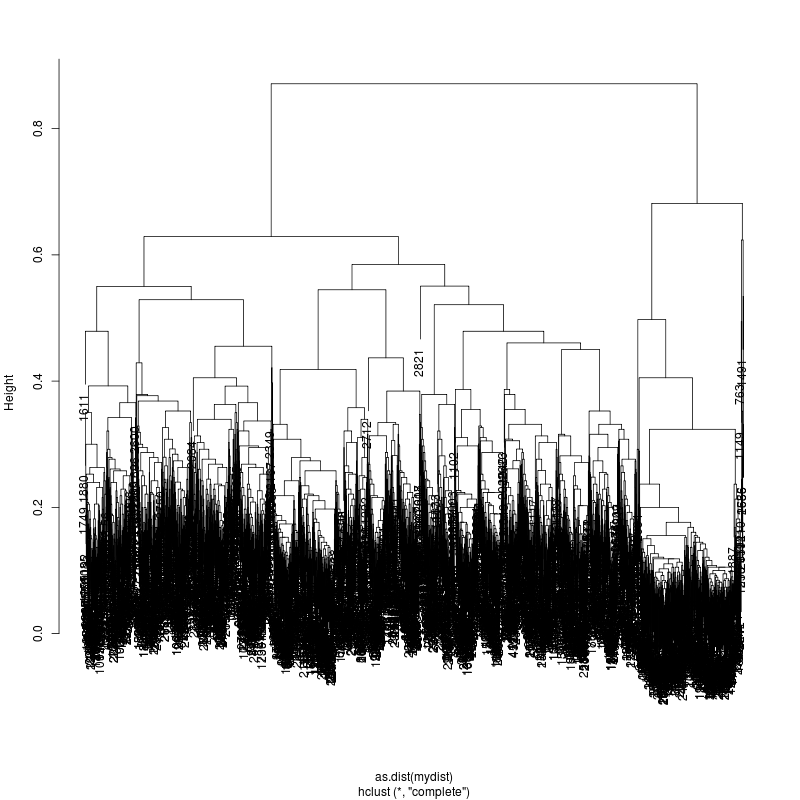
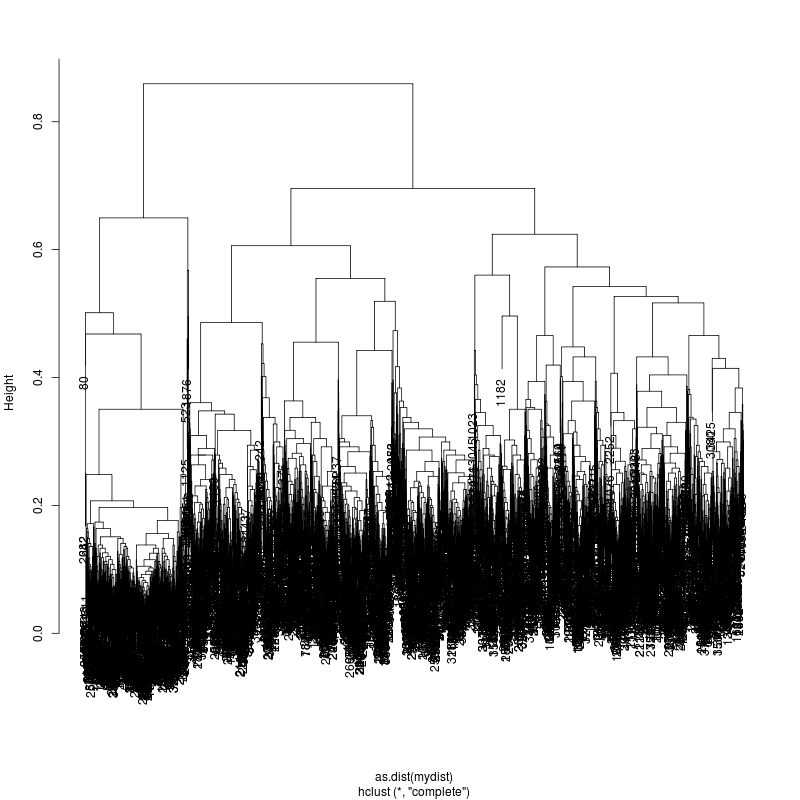 20MIX
20MIX
 - I started with the 20 genome mix everything minus 6, 13, 14, and 15
- I ran an initial deconvolution using untuned code on the 20mixes _1
and _2 and got these genomes:
20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta
20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta
There are some differences so I didn't get the same answer each
time. But this is untuned
- 11 of the 20 reported genomes are exactly the same between runs 1 and 2.
- The remaining have low comparison error (2/10,000) except for a
handful.
- The bounds say this should work and I'm sure the tuning will yield
consistent correct answers. I want to guard against getting to the
moon by climbing the tallest tree.
================================================================
Based on Jan10,2014 email here are the SGAs from a single patient (2 time points)
>>>>>>>>>>>>>>>>>>>>>>>
Total DNA
HIV-RNA Total CD4 MiSeq MiSeq amount
Sample ID Year (copies/ml) (cells/µl) analysis notes (approximate)
1 Pt3_#1 1995 191,837 32 Single 3,865 (ng)
2 Pt3_#2 1995 191,837 32 Single 1,220 (ng)
3 Pt3_#3 1995 191,837 32 Single 1,152 (ng)
4 Pt3_#4 1995 191,837 32 Single 324 (ng)
5 Pt3_#5 1995 191,837 32 Single 1,400 (ng)
6 Pt3_#6 1995 191,837 32 Single? Possible 4% contamination 3,915 (ng)
7 Pt3_#7 1995 191,837 32 Single 1,652 (ng)
8 Pt3_#8 1995 191,837 32 Single 182 (ng)
9 Pt3_#9 1995 191,837 32 Single 782 (ng)
10 Pt3_#10 1995 191,837 32 Single 870 (ng)
11 Pt3_#11 1995 191,837 32 Single 2,975 (ng)
12 Pt3_#12 1995 191,837 32 Single 2,580 (ng)
13 Pt3_#13 1995 191,837 32 Multiple Estimate of 2 375 (ng)
14 Pt3_#14 1995 191,837 32 Single 127 (ng)
15 Pt3_#15 1995 191,837 32 Multiple Estimate of 3 354 (ng)
16 Pt3_#16 1995 191,837 32 Single 408 (ng)
17 Pt3_#17 1995 191,837 32 Single 723 (ng)
18 Pt3_#18 1995 191,837 32 Single 295 (ng)
19 Pt3_#19 2001 <50 879 Single 3,596 (ng)
20 Pt3_#20 2001 <50 879 Single 2,601 (ng)
21 Pt3_#21 2001 <50 879 Single 2,240 (ng)
22 Pt3_#22 2001 <50 879 Single 1,358 (ng)
23 Pt3_#23 2001 <50 879 Single 2,756 (ng)
24 Pt3_#24 2001 <50 879 Single 1,597 (ng)
<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
Here are three sequencing runs based on these samples:
PLATE 299 in LISA started 1/15/14
Library Name sample Polymerase to template ratio onChip Conc. (pM) Well P1 Data Location Instrument Movie StageStart Chemistry Magbead
Pt3_20Mix_10pM all samples minus 6, 13, 14, and 15 2 to 1 10 A01_1, A01_2 43%, 43% \\usmp-data3\vol53\LIMS\2014\1\2014-01-15_sherri_299__NIAID_single_genome_mixes Beta2 180 Y P4C2 Y
Pt3_95_13MIX_10pM 1995 samples 1, 2, 3, 4, 5, 7, (no 8) 9, 10, 11, 12 ,16, 17, 18 2 to 1 10 B01_1, B01_2 48%, 46% \\usmp-data3\vol53\LIMS\2014\1\2014-01-15_sherri_299__NIAID_single_genome_mixes Beta2 180 Y P4C2 Y
Pt3_01_06MIX_15pM 2001 samples 19, 20, 21, 22, 23, 24 2 to 1 15 C01_1, C01_2 39%, 41% \\usmp-data3\vol53\LIMS\2014\1\2014-01-15_sherri_299__NIAID_single_genome_mixes Beta2 180 Y P4C2 Y
================================================================
So do a run against
/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/
import glob
for (name, dir) in [("Pt3_20Mix_10pM_a01_1", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/A01_1/"), ("Pt3_20Mix_10pM_a01_2", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/A01_2/"), ("Pt3_95_13MIX_10pM_B01_1", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/B01_1/"), ("Pt3_95_13MIX_10pM_B01_2", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/B01_2/"), ("Pt3_01_06MIX_15pm_C01_1", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/C01_1/"), ("Pt3_01_06MIX_15pm_C01_2", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/C01_2/")]:
# get the bas.h5 path
mybash5 = glob.glob("%s/Analysis_Results/*.bas.h5" % dir)[0]
fp = open("doit.%s.sh" % name,"w")
fp.write("cd /home/UNIXHOME/mbrown/mbrown/workspace2014Q1/niad-20mix\n")
fp.write("export SEYMOUR_HOME=/mnt/secondary/Smrtanalysis/opt/smrtanalysis; source $SEYMOUR_HOME/etc/setup.sh;\n")
fp.write("export PATH=/home/UNIXHOME/mbrown/mbrown/workspace2013Q1/pacbioCode-viral-clusteringConsensus-v1/code:$PATH\n")
fp.write("ls %s/Analysis_Results/*.bax.h5 > %s.baxh5.fofn\n" % (dir, name))
fp.write("bash5tools.py --outFilePrefix raw-niaid-%s --readType unrolled --outType fasta --minLength 2048 %s\n" % (name, mybash5))
fp.write("time ConsensusClusterSubset.py --nproc=8 --runDir=clucon-%s --fasta=raw-niaid-%s.fasta --ref=/home/UNIXHOME/mbrown/mbrown/workspace2013Q4/niaid-hiv-analysis/hiv_hxb2_whole_genome-covered.fasta --spanThreshold=99.0%% --entropyThreshold=1.0 --basfofn=%s.baxh5.fofn > %s.output 2>&1\n" % (name,name,name,name))
fp.close()
print "qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.%s.sh" % name
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_20Mix_10pM_a01_1.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_20Mix_10pM_a01_2.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_95_13MIX_10pM_B01_1.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_95_13MIX_10pM_B01_2.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_01_06MIX_15pm_C01_1.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_01_06MIX_15pm_C01_2.sh
================================================================
---- Number of reads:
ls clucon*/alignments.filterFull | grep -v "D1C" | xargs -i wc {}
3387 23709 489131 clucon-Pt3_01_06MIX_15pm_C01_1/alignments.filterFull
2788 19516 402953 clucon-Pt3_01_06MIX_15pm_C01_2/alignments.filterFull
3801 26607 549209 clucon-Pt3_20Mix_10pM_a01_1/alignments.filterFull
3607 25249 521044 clucon-Pt3_20Mix_10pM_a01_2/alignments.filterFull
3030 21210 437524 clucon-Pt3_95_13MIX_10pM_B01_1/alignments.filterFull
3559 24913 514262 clucon-Pt3_95_13MIX_10pM_B01_2/alignments.filterFull
About 3000 full length reads per chip.
---- Clustering trees:
ls -1 clucon*/aac.hclust.png | grep -v "_D"
6MIX
13MIX
20MIX
================================================================
Now all are done.
Cut the tree such that there are K groups each with at least 50 sequences in each group.
Start in clucon-Pt3_20Mix_10pM_a01_1
myct = cutree(myhc, k=20)
sum(table(myct)>50)
[1] 16
too few...
myct = cutree(myhc, k=23)
sum(table(myct)>50)
[1] 19
sort(table(myct))
20 23 22 21 19 9 12 2 4 8 14 11 18 16 15 17 6 10 13 3
1 1 3 4 96 100 103 111 117 146 148 152 158 160 163 174 187 232 276 304
5 1 7
334 377 454
myct = cutree(myhc, k=24)
sum(table(myct)>50)
[1] 19
sort(table(myct))
20 24 22 21 23 19 9 12 2 4 8 14 11 18 16 15 17 6 10 13
1 1 3 4 4 92 100 103 111 117 146 148 152 158 160 163 174 187 232 276
3 5 1 7
304 334 377 454
myct = cutree(myhc, k=25)
sum(table(myct)>50)
[1] 20
sort(table(myct))
21 25 23 22 24 20 9 13 2 4 8 15 12 19 17 1 16 18 6 10
1 1 3 4 4 92 100 103 111 117 146 148 152 158 160 162 163 174 187 215
11 14 3 5 7
232 276 304 334 454
myct = cutree(myhc, k=26)
sum(table(myct)>50)
[1] 20
sort(table(myct))
21 24 26 23 22 25 20 9 13 2 4 8 15 12 19 17 1 16 18 6
1 1 1 3 4 4 92 100 103 111 117 146 148 152 158 159 162 163 174 187
10 11 14 3 5 7
215 232 276 304 334 454
myct = cutree(myhc, k=27)
sum(table(myct)>50)
[1] 20
sort(table(myct))
21 25 27 24 22 26 23 20 9 13 2 4 8 15 12 19 17 1 16 18
1 1 1 3 4 4 6 92 94 103 111 117 146 148 152 158 159 162 163 174
6 10 11 14 3 5 7
187 215 232 276 304 334 454
myct = cutree(myhc, k=28)
sum(table(myct)>50)
[1] 21
sort(table(myct))
22 26 28 25 23 27 24 21 9 13 2 4 16 8 15 12 20 18 1 17
1 1 1 3 4 4 6 92 94 103 111 117 120 146 148 152 158 159 162 163
19 3 6 10 11 14 5 7
174 184 187 215 232 276 334 454
myct = cutree(myhc, k=29)
sum(table(myct)>50)
[1] 21
sort(table(myct))
myct
22 26 27 29 25 23 28 24 21 9 13 2 4 16 8 15 12 20 18 1
1 1 1 1 3 4 4 6 92 94 103 111 117 120 146 148 151 158 159 162
17 19 3 6 10 11 14 5 7
163 174 184 187 215 232 276 334 454
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
>>> try the k=25 cut
myct = cutree(myhc, k=25)
myid = read.table("aac.id", head=F,sep="\t")
# the first sequence is always the reference the way I'm running
# it. And get rid of the subread information
jj = cbind(as.character(gsub("\\/[0-9_]+$","",myid$V1[2:length(myct)])), myct[2:length(myct)])
writegroup = function(group,name){
file = paste("clustergroup_",name,".ids",sep="")
cat(jj[ jj[,2]==group, 1], file=file, sep="\n")
}
bysize = names(sort(table(jj[,2]),decr=T))
for (ii in 1:length(bysize)){
writegroup(bysize[ii],ii)
}
delete the less than 92 groups (removed 5 clustergroup_21.ids : clustergroup_25.ids)
Recurse:
export SEYMOUR_HOME=/mnt/secondary/Smrtanalysis/opt/smrtanalysis; source $SEYMOUR_HOME/etc/setup.sh;
export PATH=/home/UNIXHOME/mbrown/mbrown/workspace2013Q1/pacbioCode-viral-clusteringConsensus-v1/code:$PATH
cluconRecurse.py clucon-Pt3_20Mix_10pM_a01_1 > clucon-Pt3_20Mix_10pM_a01_1.cluconRecurse
grep export clucon-Pt3_20Mix_10pM_a01_1.cluconRecurse | python /home/UNIXHOME/mbrown/mbrown/workspace2013Q3/partners-hiv/qsubit.py
import glob
fp=open("20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta","w")
ggg = glob.glob("clucon-Pt3_20Mix_10pM_a01_1-~_*/quiverResult.consensus.fasta")
c=0
for gg in ggg:
dat = open(gg).read().splitlines()
fp.write(">20gs-clucon-Pt3_20Mix_10pM_a01_1-num%d\n" % c)
c+=1
for ii in range(1,len(dat)):
fp.write("%s\n" % dat[ii])
fp.close()
20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta
RESULT: these are the 20 consensus genomes for run
clucon-Pt3_20Mix_10pM_a01_1
================================================================
================================================================
Now try clucon-Pt3_20Mix_10pM_a01_2 to see if I get the same 20.
myct = cutree(myhc, k=23)
sum(table(myct)>50)
[1] 18
sort(table(myct))
2 2 2 3 7 70 96 136 140 144 168 169 171 172 177 188 199 203 206 262
2 4 14
323 352 415
24 = 18
25 = 18
26 = 18
27 = 19
myct = cutree(myhc, k=28)
sum(table(myct)>50)
[1] 20
sort(table(myct))
25 27 22 26 28 24 20 23 19 10 18 21 6 9 3 11 2 17 5 8
1 1 2 2 2 3 4 7 70 91 96 108 132 140 144 159 164 168 169 170
16 1 14 7 13 12 4 15
171 177 188 203 206 262 352 415
myct = cutree(myhc, k=29)
sum(table(myct)>50)
[1] 20
sort(table(myct))
26 28 22 25 27 29 24 20 23 19 10 18 21 6 9 3 11 2 17 5
1 1 2 2 2 2 3 4 7 70 91 96 108 132 140 142 159 164 168 169
8 16 1 14 7 13 12 4 15
170 171 177 188 203 206 262 352 415
myct = cutree(myhc, k=35)
sum(table(myct)>50)
[1] 21
29 30 31 32 33 34 23 24 27 28 35 21 25 26 20 15 10 19 1 22
1 1 1 1 1 1 2 2 2 2 2 4 7 10 70 80 91 96 97 98
6 9 3 11 2 5 18 8 17 14 7 13 12 4 16
130 140 142 159 164 168 168 170 171 188 203 206 262 352 415
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> Try the k=28 cut
myct = cutree(myhc, k=28)
myid = read.table("aac.id", head=F,sep="\t")
# the first sequence is always the reference the way I'm running
# it. And get rid of the subread information
jj = cbind(as.character(gsub("\\/[0-9_]+$","",myid$V1[2:length(myct)])), myct[2:length(myct)])
writegroup = function(group,name){
file = paste("clustergroup_",name,".ids",sep="")
cat(jj[ jj[,2]==group, 1], file=file, sep="\n")
}
bysize = names(sort(table(jj[,2]),decr=T))
for (ii in 1:length(bysize)){
writegroup(bysize[ii],ii)
}
delete the less than 70 groups (removed 8 clustergroup_21.ids : clustergroup_28.ids)
Recurse:
export SEYMOUR_HOME=/mnt/secondary/Smrtanalysis/opt/smrtanalysis; source $SEYMOUR_HOME/etc/setup.sh;
export PATH=/home/UNIXHOME/mbrown/mbrown/workspace2013Q1/pacbioCode-viral-clusteringConsensus-v1/code:$PATH
cluconRecurse.py clucon-Pt3_20Mix_10pM_a01_2 > clucon-Pt3_20Mix_10pM_a01_2.cluconRecurse
grep export clucon-Pt3_20Mix_10pM_a01_2.cluconRecurse | python /home/UNIXHOME/mbrown/mbrown/workspace2013Q3/partners-hiv/qsubit.py
import glob
fp=open("20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta","w")
ggg = glob.glob("clucon-Pt3_20Mix_10pM_a01_2-~_*/quiverResult.consensus.fasta")
c=0
for gg in ggg:
dat = open(gg).read().splitlines()
fp.write(">20gs-clucon-Pt3_20Mix_10pM_a01_2-num%d\n" % c)
c+=1
for ii in range(1,len(dat)):
fp.write("%s\n" % dat[ii])
fp.close()
20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta
RESULT: these are the 20 consensus genomes for run
clucon-Pt3_20Mix_10pM_a01_2
Looking at the file sizes, I see some inconsistency. For size=9176 _1
has 4 but _2 has 3. I would not expect this if we got the same answer
each time. The size of the total genomes is about 250 bases different!
So I can't be getting the same answers.
================================================================
================================================================
================================================================
Compare all the genomes by aligning using blasr and sorting on aligment score
export SEYMOUR_HOME=/mnt/secondary/Smrtanalysis/opt/smrtanalysis; source $SEYMOUR_HOME/etc/setup.sh;
export PATH=/home/UNIXHOME/mbrown/mbrown/workspace2013Q1/pacbioCode-viral-clusteringConsensus-v1/code:$PATH
blasr 20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta 20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta > blasr_1_2.compare
blasr 20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta 20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta > blasr_1_1.compare
blasr 20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta 20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta > blasr_2_2.compare
Replace:
20gs-clucon-Pt3_20Mix_10pM_a01_1-num
20gs-clucon-Pt3_20Mix_10pM_a01_2-num
with nothing to make analysis easier.
----
dat = read.table("blasr_1_1.compare", head=F,sep=" ")
iden = dat[dat$V6==100.0,]
V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 V11 V12 V13
1 0 0 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
2 0 5 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
6 1 1 0 0 -44835 100 0 8967 8967 0 8967 8967 188275
8 2 2 0 0 -44895 100 0 8979 8979 0 8979 8979 188527
14 3 3 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
19 4 4 0 0 -44840 100 0 8968 8968 0 8968 8968 188296
21 5 0 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
22 5 5 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
26 6 6 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
34 7 7 0 0 -44875 100 0 8975 8975 0 8975 8975 188443
42 8 8 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
48 9 9 0 0 -44960 100 0 8992 8992 0 8992 8992 188800
55 10 10 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
56 11 11 0 0 -44815 100 0 8963 8963 0 8963 8963 188191
63 12 12 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
68 13 13 0 0 -44815 100 0 8963 8963 0 8963 8963 188191
75 14 14 0 0 -44950 100 0 8990 8990 0 8990 8990 188758
80 15 15 0 0 -41930 100 0 8386 8386 0 8386 8386 176074
85 16 16 0 0 -43040 100 0 8608 8608 0 8608 8608 180736
89 17 17 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
94 18 18 0 0 -42465 100 0 8493 8493 0 8493 8493 178321
101 19 19 0 0 -44910 100 0 8982 8982 0 8982 8982 188590
Run one genomes 0 and 5 are identical
----
dat = read.table("blasr_2_2.compare", head=F,sep=" ")
iden = dat[dat$V6==100.0,]
V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 V11 V12 V13
1 0 0 0 0 -44915 100 0 8983 8983 0 8983 8983 188611
2 1 1 0 0 -44965 100 0 8993 8993 0 8993 8993 188821
9 2 2 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
13 3 3 0 0 -41935 100 0 8387 8387 0 8387 8387 176095
17 4 4 0 0 -43050 100 0 8610 8610 0 8610 8610 180778
18 5 5 0 0 -42480 100 0 8496 8496 0 8496 8496 178384
25 6 6 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
29 7 7 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
31 8 8 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
32 9 9 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
33 9 16 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
40 10 10 0 0 -44875 100 0 8975 8975 0 8975 8975 188443
48 11 11 0 0 -44815 100 0 8963 8963 0 8963 8963 188191
54 12 12 0 0 -44895 100 0 8979 8979 0 8979 8979 188527
58 13 13 0 0 -44950 100 0 8990 8990 0 8990 8990 188758
62 14 14 0 0 -44830 100 0 8966 8966 0 8966 8966 188254
64 15 15 0 0 -43655 100 0 8731 8731 0 8731 8731 183319
73 16 9 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
74 16 16 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
81 17 17 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
87 18 18 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
91 19 19 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
Run two genomes 9 and 16 are identical
----
dat = read.table("blasr_1_2.compare", head=F,sep=" ")
Here are the identical genomes between the two runs:
iden = dat[dat$V6==100.0,]
V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 V11 V12 V13
1 0 2 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
17 5 2 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
.
21 6 9 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
22 6 16 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
.
7 2 12 0 0 -44895 100 0 8979 8979 0 8979 8979 188527
11 3 18 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
29 7 10 0 0 -44875 100 0 8975 8975 0 8975 8975 188443
37 8 6 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
48 10 8 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
54 12 19 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
60 13 11 0 0 -44815 100 0 8963 8963 0 8963 8963 188191
66 14 13 0 0 -44950 100 0 8990 8990 0 8990 8990 188758
90 17 17 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
Genome 6 in run1 is the same as genome 9 and 16 in run2.
Genome 2 in run2 is the same as genome 0 and 5 in run1.
11 genomes reported exactly the same betweeen the two runs (plus one
extra in each run yields 12 "correct" genomes).
================================
What about the other 8 geomes that aren't 100% identical.
Number from 1:
findmin1 = function(from){
tmp = dat[dat$V1==from,]
return(c(tmp$V2[which.min(tmp$V5)]+1,tmp$V6[which.min(tmp$V5)]))
}
findmin2 = function(from){
tmp = dat[dat$V2==from,]
return(c(tmp$V1[which.min(tmp$V5)]+1,tmp$V6[which.min(tmp$V5)]))
}
sapply(seq(0,19),findmin1)
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12]
[1,] 3 15.0000 13 19 15.0000 3 10 11 7 2.0000 9 10.0000
[2,] 100 99.9888 100 100 99.9777 100 100 100 100 99.9889 100 99.9888
[,13] [,14] [,15] [,16] [,17] [,18] [,19] [,20]
[1,] 20 12 14 4.0000 18.0000 18 6.0000 1.0000
[2,] 100 100 100 99.7618 94.6584 100 99.9647 99.9443
sapply(seq(0,19),findmin2)
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11]
[1,] 20.0000 10.0000 1 16.0000 12.0000 19.0000 9 2.0000 11 7 8
[2,] 99.9443 99.9889 100 99.7618 93.4749 99.9647 100 99.9665 100 100 100
[,12] [,13] [,14] [,15] [,16] [,17] [,18] [,19] [,20]
[1,] 14 3 15 2.0000 20.0000 7 18 4 13
[2,] 100 100 100 99.9888 95.4879 100 100 100 100
The imperfected mapping are 99.9x except for run1=17, run2=5,16
- I started with the 20 genome mix everything minus 6, 13, 14, and 15
- I ran an initial deconvolution using untuned code on the 20mixes _1
and _2 and got these genomes:
20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta
20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta
There are some differences so I didn't get the same answer each
time. But this is untuned
- 11 of the 20 reported genomes are exactly the same between runs 1 and 2.
- The remaining have low comparison error (2/10,000) except for a
handful.
- The bounds say this should work and I'm sure the tuning will yield
consistent correct answers. I want to guard against getting to the
moon by climbing the tallest tree.
================================================================
Based on Jan10,2014 email here are the SGAs from a single patient (2 time points)
>>>>>>>>>>>>>>>>>>>>>>>
Total DNA
HIV-RNA Total CD4 MiSeq MiSeq amount
Sample ID Year (copies/ml) (cells/µl) analysis notes (approximate)
1 Pt3_#1 1995 191,837 32 Single 3,865 (ng)
2 Pt3_#2 1995 191,837 32 Single 1,220 (ng)
3 Pt3_#3 1995 191,837 32 Single 1,152 (ng)
4 Pt3_#4 1995 191,837 32 Single 324 (ng)
5 Pt3_#5 1995 191,837 32 Single 1,400 (ng)
6 Pt3_#6 1995 191,837 32 Single? Possible 4% contamination 3,915 (ng)
7 Pt3_#7 1995 191,837 32 Single 1,652 (ng)
8 Pt3_#8 1995 191,837 32 Single 182 (ng)
9 Pt3_#9 1995 191,837 32 Single 782 (ng)
10 Pt3_#10 1995 191,837 32 Single 870 (ng)
11 Pt3_#11 1995 191,837 32 Single 2,975 (ng)
12 Pt3_#12 1995 191,837 32 Single 2,580 (ng)
13 Pt3_#13 1995 191,837 32 Multiple Estimate of 2 375 (ng)
14 Pt3_#14 1995 191,837 32 Single 127 (ng)
15 Pt3_#15 1995 191,837 32 Multiple Estimate of 3 354 (ng)
16 Pt3_#16 1995 191,837 32 Single 408 (ng)
17 Pt3_#17 1995 191,837 32 Single 723 (ng)
18 Pt3_#18 1995 191,837 32 Single 295 (ng)
19 Pt3_#19 2001 <50 879 Single 3,596 (ng)
20 Pt3_#20 2001 <50 879 Single 2,601 (ng)
21 Pt3_#21 2001 <50 879 Single 2,240 (ng)
22 Pt3_#22 2001 <50 879 Single 1,358 (ng)
23 Pt3_#23 2001 <50 879 Single 2,756 (ng)
24 Pt3_#24 2001 <50 879 Single 1,597 (ng)
<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<<
Here are three sequencing runs based on these samples:
PLATE 299 in LISA started 1/15/14
Library Name sample Polymerase to template ratio onChip Conc. (pM) Well P1 Data Location Instrument Movie StageStart Chemistry Magbead
Pt3_20Mix_10pM all samples minus 6, 13, 14, and 15 2 to 1 10 A01_1, A01_2 43%, 43% \\usmp-data3\vol53\LIMS\2014\1\2014-01-15_sherri_299__NIAID_single_genome_mixes Beta2 180 Y P4C2 Y
Pt3_95_13MIX_10pM 1995 samples 1, 2, 3, 4, 5, 7, (no 8) 9, 10, 11, 12 ,16, 17, 18 2 to 1 10 B01_1, B01_2 48%, 46% \\usmp-data3\vol53\LIMS\2014\1\2014-01-15_sherri_299__NIAID_single_genome_mixes Beta2 180 Y P4C2 Y
Pt3_01_06MIX_15pM 2001 samples 19, 20, 21, 22, 23, 24 2 to 1 15 C01_1, C01_2 39%, 41% \\usmp-data3\vol53\LIMS\2014\1\2014-01-15_sherri_299__NIAID_single_genome_mixes Beta2 180 Y P4C2 Y
================================================================
So do a run against
/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/
import glob
for (name, dir) in [("Pt3_20Mix_10pM_a01_1", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/A01_1/"), ("Pt3_20Mix_10pM_a01_2", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/A01_2/"), ("Pt3_95_13MIX_10pM_B01_1", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/B01_1/"), ("Pt3_95_13MIX_10pM_B01_2", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/B01_2/"), ("Pt3_01_06MIX_15pm_C01_1", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/C01_1/"), ("Pt3_01_06MIX_15pm_C01_2", "/mnt/data3/vol53/LIMS/2014/1/2014-01-15_sherri_299__NIAID_single_genome_mixes/C01_2/")]:
# get the bas.h5 path
mybash5 = glob.glob("%s/Analysis_Results/*.bas.h5" % dir)[0]
fp = open("doit.%s.sh" % name,"w")
fp.write("cd /home/UNIXHOME/mbrown/mbrown/workspace2014Q1/niad-20mix\n")
fp.write("export SEYMOUR_HOME=/mnt/secondary/Smrtanalysis/opt/smrtanalysis; source $SEYMOUR_HOME/etc/setup.sh;\n")
fp.write("export PATH=/home/UNIXHOME/mbrown/mbrown/workspace2013Q1/pacbioCode-viral-clusteringConsensus-v1/code:$PATH\n")
fp.write("ls %s/Analysis_Results/*.bax.h5 > %s.baxh5.fofn\n" % (dir, name))
fp.write("bash5tools.py --outFilePrefix raw-niaid-%s --readType unrolled --outType fasta --minLength 2048 %s\n" % (name, mybash5))
fp.write("time ConsensusClusterSubset.py --nproc=8 --runDir=clucon-%s --fasta=raw-niaid-%s.fasta --ref=/home/UNIXHOME/mbrown/mbrown/workspace2013Q4/niaid-hiv-analysis/hiv_hxb2_whole_genome-covered.fasta --spanThreshold=99.0%% --entropyThreshold=1.0 --basfofn=%s.baxh5.fofn > %s.output 2>&1\n" % (name,name,name,name))
fp.close()
print "qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.%s.sh" % name
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_20Mix_10pM_a01_1.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_20Mix_10pM_a01_2.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_95_13MIX_10pM_B01_1.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_95_13MIX_10pM_B01_2.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_01_06MIX_15pm_C01_1.sh
qsub -q secondary -S /bin/bash -pe smp 8 -V -cwd doit.Pt3_01_06MIX_15pm_C01_2.sh
================================================================
---- Number of reads:
ls clucon*/alignments.filterFull | grep -v "D1C" | xargs -i wc {}
3387 23709 489131 clucon-Pt3_01_06MIX_15pm_C01_1/alignments.filterFull
2788 19516 402953 clucon-Pt3_01_06MIX_15pm_C01_2/alignments.filterFull
3801 26607 549209 clucon-Pt3_20Mix_10pM_a01_1/alignments.filterFull
3607 25249 521044 clucon-Pt3_20Mix_10pM_a01_2/alignments.filterFull
3030 21210 437524 clucon-Pt3_95_13MIX_10pM_B01_1/alignments.filterFull
3559 24913 514262 clucon-Pt3_95_13MIX_10pM_B01_2/alignments.filterFull
About 3000 full length reads per chip.
---- Clustering trees:
ls -1 clucon*/aac.hclust.png | grep -v "_D"
6MIX
13MIX
20MIX
================================================================
Now all are done.
Cut the tree such that there are K groups each with at least 50 sequences in each group.
Start in clucon-Pt3_20Mix_10pM_a01_1
myct = cutree(myhc, k=20)
sum(table(myct)>50)
[1] 16
too few...
myct = cutree(myhc, k=23)
sum(table(myct)>50)
[1] 19
sort(table(myct))
20 23 22 21 19 9 12 2 4 8 14 11 18 16 15 17 6 10 13 3
1 1 3 4 96 100 103 111 117 146 148 152 158 160 163 174 187 232 276 304
5 1 7
334 377 454
myct = cutree(myhc, k=24)
sum(table(myct)>50)
[1] 19
sort(table(myct))
20 24 22 21 23 19 9 12 2 4 8 14 11 18 16 15 17 6 10 13
1 1 3 4 4 92 100 103 111 117 146 148 152 158 160 163 174 187 232 276
3 5 1 7
304 334 377 454
myct = cutree(myhc, k=25)
sum(table(myct)>50)
[1] 20
sort(table(myct))
21 25 23 22 24 20 9 13 2 4 8 15 12 19 17 1 16 18 6 10
1 1 3 4 4 92 100 103 111 117 146 148 152 158 160 162 163 174 187 215
11 14 3 5 7
232 276 304 334 454
myct = cutree(myhc, k=26)
sum(table(myct)>50)
[1] 20
sort(table(myct))
21 24 26 23 22 25 20 9 13 2 4 8 15 12 19 17 1 16 18 6
1 1 1 3 4 4 92 100 103 111 117 146 148 152 158 159 162 163 174 187
10 11 14 3 5 7
215 232 276 304 334 454
myct = cutree(myhc, k=27)
sum(table(myct)>50)
[1] 20
sort(table(myct))
21 25 27 24 22 26 23 20 9 13 2 4 8 15 12 19 17 1 16 18
1 1 1 3 4 4 6 92 94 103 111 117 146 148 152 158 159 162 163 174
6 10 11 14 3 5 7
187 215 232 276 304 334 454
myct = cutree(myhc, k=28)
sum(table(myct)>50)
[1] 21
sort(table(myct))
22 26 28 25 23 27 24 21 9 13 2 4 16 8 15 12 20 18 1 17
1 1 1 3 4 4 6 92 94 103 111 117 120 146 148 152 158 159 162 163
19 3 6 10 11 14 5 7
174 184 187 215 232 276 334 454
myct = cutree(myhc, k=29)
sum(table(myct)>50)
[1] 21
sort(table(myct))
myct
22 26 27 29 25 23 28 24 21 9 13 2 4 16 8 15 12 20 18 1
1 1 1 1 3 4 4 6 92 94 103 111 117 120 146 148 151 158 159 162
17 19 3 6 10 11 14 5 7
163 174 184 187 215 232 276 334 454
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
>>> try the k=25 cut
myct = cutree(myhc, k=25)
myid = read.table("aac.id", head=F,sep="\t")
# the first sequence is always the reference the way I'm running
# it. And get rid of the subread information
jj = cbind(as.character(gsub("\\/[0-9_]+$","",myid$V1[2:length(myct)])), myct[2:length(myct)])
writegroup = function(group,name){
file = paste("clustergroup_",name,".ids",sep="")
cat(jj[ jj[,2]==group, 1], file=file, sep="\n")
}
bysize = names(sort(table(jj[,2]),decr=T))
for (ii in 1:length(bysize)){
writegroup(bysize[ii],ii)
}
delete the less than 92 groups (removed 5 clustergroup_21.ids : clustergroup_25.ids)
Recurse:
export SEYMOUR_HOME=/mnt/secondary/Smrtanalysis/opt/smrtanalysis; source $SEYMOUR_HOME/etc/setup.sh;
export PATH=/home/UNIXHOME/mbrown/mbrown/workspace2013Q1/pacbioCode-viral-clusteringConsensus-v1/code:$PATH
cluconRecurse.py clucon-Pt3_20Mix_10pM_a01_1 > clucon-Pt3_20Mix_10pM_a01_1.cluconRecurse
grep export clucon-Pt3_20Mix_10pM_a01_1.cluconRecurse | python /home/UNIXHOME/mbrown/mbrown/workspace2013Q3/partners-hiv/qsubit.py
import glob
fp=open("20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta","w")
ggg = glob.glob("clucon-Pt3_20Mix_10pM_a01_1-~_*/quiverResult.consensus.fasta")
c=0
for gg in ggg:
dat = open(gg).read().splitlines()
fp.write(">20gs-clucon-Pt3_20Mix_10pM_a01_1-num%d\n" % c)
c+=1
for ii in range(1,len(dat)):
fp.write("%s\n" % dat[ii])
fp.close()
20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta
RESULT: these are the 20 consensus genomes for run
clucon-Pt3_20Mix_10pM_a01_1
================================================================
================================================================
Now try clucon-Pt3_20Mix_10pM_a01_2 to see if I get the same 20.
myct = cutree(myhc, k=23)
sum(table(myct)>50)
[1] 18
sort(table(myct))
2 2 2 3 7 70 96 136 140 144 168 169 171 172 177 188 199 203 206 262
2 4 14
323 352 415
24 = 18
25 = 18
26 = 18
27 = 19
myct = cutree(myhc, k=28)
sum(table(myct)>50)
[1] 20
sort(table(myct))
25 27 22 26 28 24 20 23 19 10 18 21 6 9 3 11 2 17 5 8
1 1 2 2 2 3 4 7 70 91 96 108 132 140 144 159 164 168 169 170
16 1 14 7 13 12 4 15
171 177 188 203 206 262 352 415
myct = cutree(myhc, k=29)
sum(table(myct)>50)
[1] 20
sort(table(myct))
26 28 22 25 27 29 24 20 23 19 10 18 21 6 9 3 11 2 17 5
1 1 2 2 2 2 3 4 7 70 91 96 108 132 140 142 159 164 168 169
8 16 1 14 7 13 12 4 15
170 171 177 188 203 206 262 352 415
myct = cutree(myhc, k=35)
sum(table(myct)>50)
[1] 21
29 30 31 32 33 34 23 24 27 28 35 21 25 26 20 15 10 19 1 22
1 1 1 1 1 1 2 2 2 2 2 4 7 10 70 80 91 96 97 98
6 9 3 11 2 5 18 8 17 14 7 13 12 4 16
130 140 142 159 164 168 168 170 171 188 203 206 262 352 415
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
> Try the k=28 cut
myct = cutree(myhc, k=28)
myid = read.table("aac.id", head=F,sep="\t")
# the first sequence is always the reference the way I'm running
# it. And get rid of the subread information
jj = cbind(as.character(gsub("\\/[0-9_]+$","",myid$V1[2:length(myct)])), myct[2:length(myct)])
writegroup = function(group,name){
file = paste("clustergroup_",name,".ids",sep="")
cat(jj[ jj[,2]==group, 1], file=file, sep="\n")
}
bysize = names(sort(table(jj[,2]),decr=T))
for (ii in 1:length(bysize)){
writegroup(bysize[ii],ii)
}
delete the less than 70 groups (removed 8 clustergroup_21.ids : clustergroup_28.ids)
Recurse:
export SEYMOUR_HOME=/mnt/secondary/Smrtanalysis/opt/smrtanalysis; source $SEYMOUR_HOME/etc/setup.sh;
export PATH=/home/UNIXHOME/mbrown/mbrown/workspace2013Q1/pacbioCode-viral-clusteringConsensus-v1/code:$PATH
cluconRecurse.py clucon-Pt3_20Mix_10pM_a01_2 > clucon-Pt3_20Mix_10pM_a01_2.cluconRecurse
grep export clucon-Pt3_20Mix_10pM_a01_2.cluconRecurse | python /home/UNIXHOME/mbrown/mbrown/workspace2013Q3/partners-hiv/qsubit.py
import glob
fp=open("20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta","w")
ggg = glob.glob("clucon-Pt3_20Mix_10pM_a01_2-~_*/quiverResult.consensus.fasta")
c=0
for gg in ggg:
dat = open(gg).read().splitlines()
fp.write(">20gs-clucon-Pt3_20Mix_10pM_a01_2-num%d\n" % c)
c+=1
for ii in range(1,len(dat)):
fp.write("%s\n" % dat[ii])
fp.close()
20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta
RESULT: these are the 20 consensus genomes for run
clucon-Pt3_20Mix_10pM_a01_2
Looking at the file sizes, I see some inconsistency. For size=9176 _1
has 4 but _2 has 3. I would not expect this if we got the same answer
each time. The size of the total genomes is about 250 bases different!
So I can't be getting the same answers.
================================================================
================================================================
================================================================
Compare all the genomes by aligning using blasr and sorting on aligment score
export SEYMOUR_HOME=/mnt/secondary/Smrtanalysis/opt/smrtanalysis; source $SEYMOUR_HOME/etc/setup.sh;
export PATH=/home/UNIXHOME/mbrown/mbrown/workspace2013Q1/pacbioCode-viral-clusteringConsensus-v1/code:$PATH
blasr 20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta 20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta > blasr_1_2.compare
blasr 20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta 20genomes-clucon-Pt3_20Mix_10pM_a01_1.fasta > blasr_1_1.compare
blasr 20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta 20genomes-clucon-Pt3_20Mix_10pM_a01_2.fasta > blasr_2_2.compare
Replace:
20gs-clucon-Pt3_20Mix_10pM_a01_1-num
20gs-clucon-Pt3_20Mix_10pM_a01_2-num
with nothing to make analysis easier.
----
dat = read.table("blasr_1_1.compare", head=F,sep=" ")
iden = dat[dat$V6==100.0,]
V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 V11 V12 V13
1 0 0 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
2 0 5 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
6 1 1 0 0 -44835 100 0 8967 8967 0 8967 8967 188275
8 2 2 0 0 -44895 100 0 8979 8979 0 8979 8979 188527
14 3 3 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
19 4 4 0 0 -44840 100 0 8968 8968 0 8968 8968 188296
21 5 0 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
22 5 5 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
26 6 6 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
34 7 7 0 0 -44875 100 0 8975 8975 0 8975 8975 188443
42 8 8 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
48 9 9 0 0 -44960 100 0 8992 8992 0 8992 8992 188800
55 10 10 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
56 11 11 0 0 -44815 100 0 8963 8963 0 8963 8963 188191
63 12 12 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
68 13 13 0 0 -44815 100 0 8963 8963 0 8963 8963 188191
75 14 14 0 0 -44950 100 0 8990 8990 0 8990 8990 188758
80 15 15 0 0 -41930 100 0 8386 8386 0 8386 8386 176074
85 16 16 0 0 -43040 100 0 8608 8608 0 8608 8608 180736
89 17 17 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
94 18 18 0 0 -42465 100 0 8493 8493 0 8493 8493 178321
101 19 19 0 0 -44910 100 0 8982 8982 0 8982 8982 188590
Run one genomes 0 and 5 are identical
----
dat = read.table("blasr_2_2.compare", head=F,sep=" ")
iden = dat[dat$V6==100.0,]
V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 V11 V12 V13
1 0 0 0 0 -44915 100 0 8983 8983 0 8983 8983 188611
2 1 1 0 0 -44965 100 0 8993 8993 0 8993 8993 188821
9 2 2 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
13 3 3 0 0 -41935 100 0 8387 8387 0 8387 8387 176095
17 4 4 0 0 -43050 100 0 8610 8610 0 8610 8610 180778
18 5 5 0 0 -42480 100 0 8496 8496 0 8496 8496 178384
25 6 6 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
29 7 7 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
31 8 8 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
32 9 9 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
33 9 16 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
40 10 10 0 0 -44875 100 0 8975 8975 0 8975 8975 188443
48 11 11 0 0 -44815 100 0 8963 8963 0 8963 8963 188191
54 12 12 0 0 -44895 100 0 8979 8979 0 8979 8979 188527
58 13 13 0 0 -44950 100 0 8990 8990 0 8990 8990 188758
62 14 14 0 0 -44830 100 0 8966 8966 0 8966 8966 188254
64 15 15 0 0 -43655 100 0 8731 8731 0 8731 8731 183319
73 16 9 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
74 16 16 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
81 17 17 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
87 18 18 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
91 19 19 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
Run two genomes 9 and 16 are identical
----
dat = read.table("blasr_1_2.compare", head=F,sep=" ")
Here are the identical genomes between the two runs:
iden = dat[dat$V6==100.0,]
V1 V2 V3 V4 V5 V6 V7 V8 V9 V10 V11 V12 V13
1 0 2 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
17 5 2 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
.
21 6 9 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
22 6 16 0 0 -44820 100 0 8964 8964 0 8964 8964 188212
.
7 2 12 0 0 -44895 100 0 8979 8979 0 8979 8979 188527
11 3 18 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
29 7 10 0 0 -44875 100 0 8975 8975 0 8975 8975 188443
37 8 6 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
48 10 8 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
54 12 19 0 0 -44890 100 0 8978 8978 0 8978 8978 188506
60 13 11 0 0 -44815 100 0 8963 8963 0 8963 8963 188191
66 14 13 0 0 -44950 100 0 8990 8990 0 8990 8990 188758
90 17 17 0 0 -44905 100 0 8981 8981 0 8981 8981 188569
Genome 6 in run1 is the same as genome 9 and 16 in run2.
Genome 2 in run2 is the same as genome 0 and 5 in run1.
11 genomes reported exactly the same betweeen the two runs (plus one
extra in each run yields 12 "correct" genomes).
================================
What about the other 8 geomes that aren't 100% identical.
Number from 1:
findmin1 = function(from){
tmp = dat[dat$V1==from,]
return(c(tmp$V2[which.min(tmp$V5)]+1,tmp$V6[which.min(tmp$V5)]))
}
findmin2 = function(from){
tmp = dat[dat$V2==from,]
return(c(tmp$V1[which.min(tmp$V5)]+1,tmp$V6[which.min(tmp$V5)]))
}
sapply(seq(0,19),findmin1)
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12]
[1,] 3 15.0000 13 19 15.0000 3 10 11 7 2.0000 9 10.0000
[2,] 100 99.9888 100 100 99.9777 100 100 100 100 99.9889 100 99.9888
[,13] [,14] [,15] [,16] [,17] [,18] [,19] [,20]
[1,] 20 12 14 4.0000 18.0000 18 6.0000 1.0000
[2,] 100 100 100 99.7618 94.6584 100 99.9647 99.9443
sapply(seq(0,19),findmin2)
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11]
[1,] 20.0000 10.0000 1 16.0000 12.0000 19.0000 9 2.0000 11 7 8
[2,] 99.9443 99.9889 100 99.7618 93.4749 99.9647 100 99.9665 100 100 100
[,12] [,13] [,14] [,15] [,16] [,17] [,18] [,19] [,20]
[1,] 14 3 15 2.0000 20.0000 7 18 4 13
[2,] 100 100 100 99.9888 95.4879 100 100 100 100
The imperfected mapping are 99.9x except for run1=17, run2=5,16